In 1991, a randomized controlled trial across 33 centers in seven countries demonstrated that folic acid reduced the recurrence of neural tube defects by 72%.¹ It was the kind of result that changes policy. By 1998, the U.S. had mandated its addition to every bag of enriched flour in the country. Neural tube defects dropped immediately, an estimated 1,326 births protected every year.² Serum folate deficiency fell from 30% of the population to less than 1%.³ No supplement in modern history has a stronger public health record. And yet, the prevailing message online in 2025 is that folic acid is dangerous, that it accumulates in your blood, that your genes can't process it, and that it should be replaced with only the active form, methylfolate. Is the most successful fortification program in modern history actually harming people?
The Conversion Bottleneck. What Happens After You Swallow It
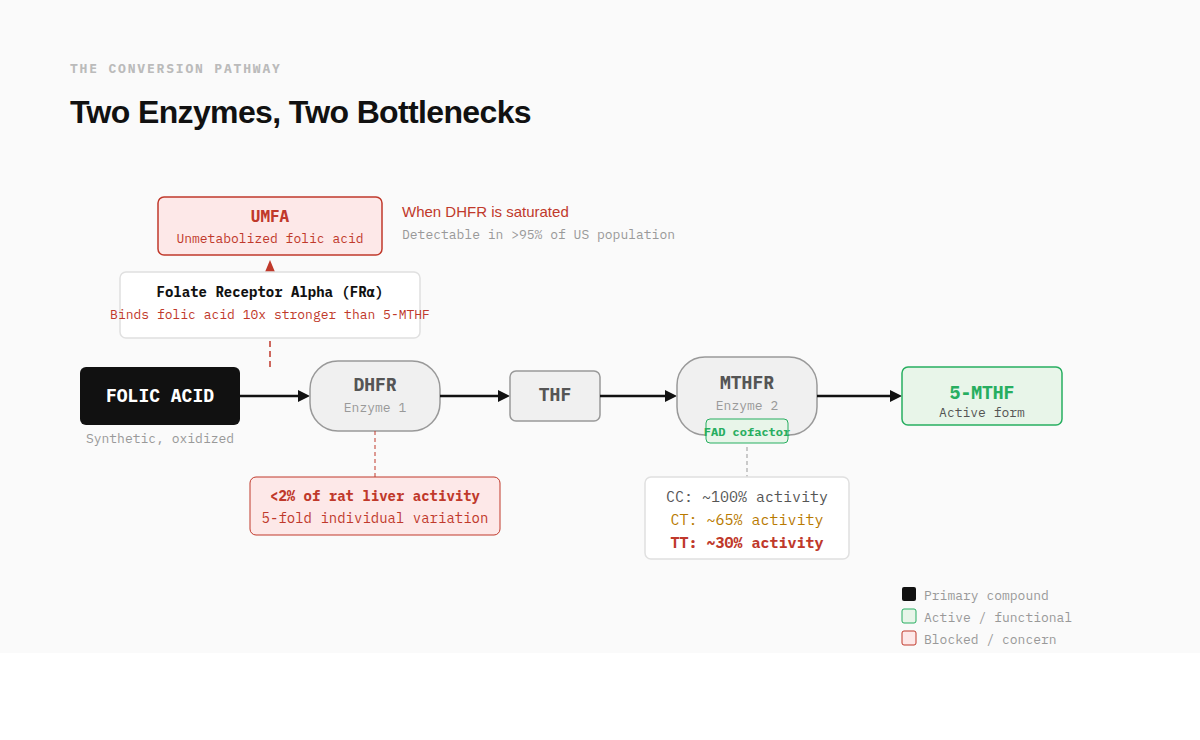
Folic acid does not exist in food. It was first synthesized in 1945 as a stable, oxidized form of vitamin B9 that could survive storage and manufacturing. The folate in spinach or lentils is already in a reduced form your body can work with. Folic acid is not. It needs to be converted (i.e., reduced) first.
That conversion happens in two steps, each controlled by a different enzyme.
The first is dihydrofolate reductase, or DHFR. This enzyme reduces folic acid into dihydrofolate, then into tetrahydrofolate. In 2009, Bailey and Ayling measured DHFR activity in fresh human liver samples and found it averaged less than 2% of the activity in rat liver, with nearly 5-fold variation between individuals.⁴ This is the bottleneck the wellness space has latched onto, and it is real. Humans convert folic acid slowly. When intake outpaces DHFR's capacity, unconverted folic acid appears in the bloodstream. This is called unmetabolized folic acid, or UMFA, and it is detectable in over 95% of the U.S. population.⁵
The second enzyme is methylenetetrahydrofolate reductase, or MTHFR. Its job is to take the tetrahydrofolate that DHFR produced and convert it into the final active form: 5-methyltetrahydrofolate. This is the form your body actually uses for DNA synthesis and homocysteine recycling. MTHFR is where the gene variants come in. The most studied and common of them, C677T, can reduce this enzyme's activity to roughly 30% in people who carry two copies of the variant (677TT). That is a meaningful reduction, and it is common enough to affect 10-15% of European populations and up to 25% of Hispanic populations.⁶
So the concern has a real biochemical basis. A synthetic compound that requires two sequential enzymes to become useful, the first of which is slow and variable in humans, the second of which is genetically impaired in a significant portion of the population. And when the system can't keep up, unmetabolized folic acid accumulates in the blood.
The question is: does that matter?
Where the Fears Come From
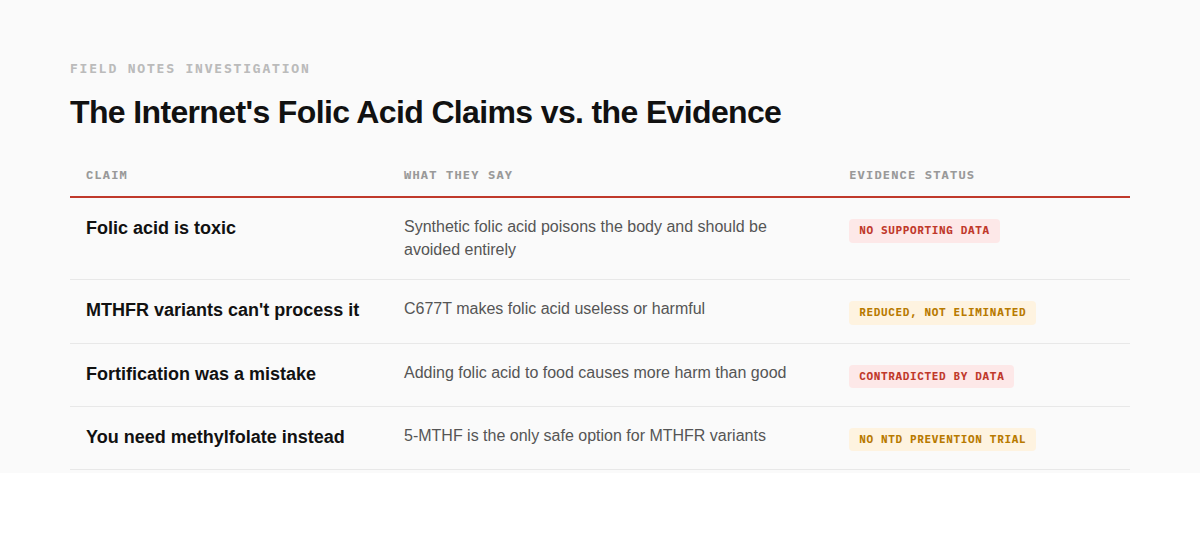
UMFA isn't just a measurement quirk. There are specific, testable reasons it might be harmful.
· Receptor Competition: UMFA may compete with natural folate (5-MTHF) for folate receptors and transporters, potentially creating a functional folate deficiency even when total blood folate looks adequate. The reason is structural: folate receptor alpha binds folic acid with roughly 10 times the affinity it has for 5-MTHF. When UMFA is circulating, it gets priority transport into cells but arrives as an unconverted compound that still needs DHFR to become functional. In tissues where DHFR activity is low, particularly the brain, the transport system may be occupied by cargo that can't be used while the usable form is locked out. Akiyama et al. (2022) demonstrated this clinically: in two patients with rare neurological conditions receiving high-dose folic acid therapy, CSF levels of 5-MTHF dropped rather than rose. Reducing or stopping the folic acid normalized them (note this was HIGH dose folic acid therapy, which does not represent the typical intakes I list below at the end of this section).⁷
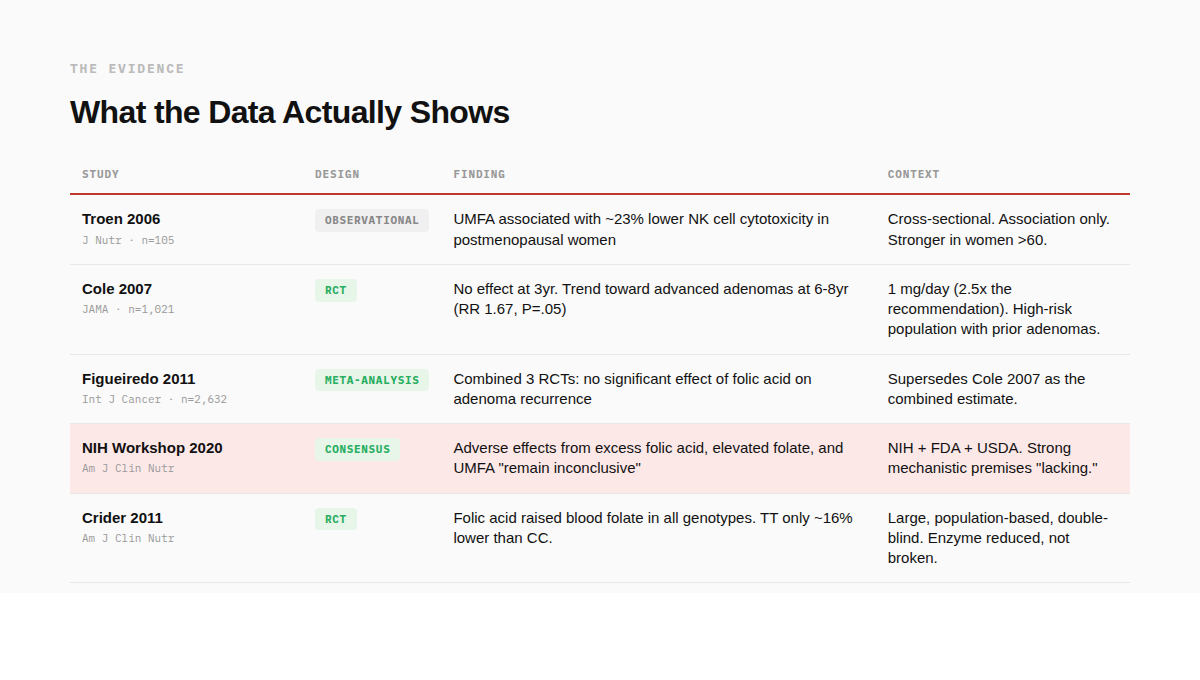
· Immune suppression. Troen et al. (2006) found that among 105 postmenopausal women, those with detectable UMFA had approximately 23% lower natural killer cell cytotoxicity, with a stronger effect in women over 60.⁸ This was observational, not causal, but NK cells are part of the immune system's surveillance against cancer and viral infection. It got attention.
· Enzyme jamming. At high concentrations, folic acid may competitively inhibit DHFR from completing its own conversion of dihydrofolate to tetrahydrofolate, creating a bottleneck on top of a bottleneck.
· B12 masking. High folate intake can correct the anemia caused by vitamin B12 deficiency while the neurological damage continues undetected. This is the original safety concern that established the tolerable upper limit at 1 mg/day.
These are not fringe claims. They come from published research and plausible pharmacology. The question is what happens when you move from mechanism to measurement.
The strongest trial-level signal is Cole et al. (2007): a double-blind, placebo-controlled study of 1,021 men and women with a history of colorectal adenomas (precancerous lesions). Participants received 1 mg per day of folic acid or placebo.
· At year 3: no difference in adenoma recurrence.
· At years 6-8: the folic acid group showed a trend toward more advanced lesions. Risk ratio 1.67 (95% CI 1.00-2.80, P = .05).⁹
This gets cited constantly, and it should be taken seriously. But two things are almost never mentioned alongside it. First, the dose was 1 mg per day, 2.5 times the standard recommendation. Second, a later combined analysis of three randomized trials (2,632 participants total) found no statistically significant effect of folic acid on adenoma recurrence or advanced lesions.¹⁰
What about the broader picture? In 2019, the NIH convened a workshop with the FDA and USDA to evaluate the full body of evidence on excess folic acid and UMFA. Their conclusion, published in 2020: adverse effects "remain inconclusive" and "do not provide the evidence needed to affect public health recommendations." They also noted that strong biological premises connecting UMFA to adverse outcomes are "lacking." ¹¹
Does Folic Acid Even Work in People with MTHFR Mutations?
Aside from the argument of safety risk, the second discussion dominating point to address is whether folic acid is even useful in someone with an MTHFR mutation – someone who has an enzyme that converts less folic acid (and/or some dietary folates) into active 5-MTHF.
Crider et al. (2011) ran a large, population-based, double-blind trial and stratified results by MTHFR C677T genotype. Folic acid raised blood folate in all three genotype groups. Individuals homozygous for the MTHFR variant (TT, associated with the lowest MTHFR activity) had levels approximately 16% lower than CC wildtype (normal enzyme function) on the same dose, but their levels still rose significantly.¹²
The enzyme is reduced, not broken. Intake compensates.
The pattern shows that harm signals emerge at supraphysiological doses (1-5 mg per day), in populations already at elevated risk, and have not been replicated in combined analyses. At the 400 mcg recommendation and the roughly 200 mcg from fortification, no trial has demonstrated harm in any population, including those with the TT genotype. The dose matters more than the genotype.
The Vitamin Nobody's Selling
The entire folic acid debate orbits one enzyme: MTHFR. Folic acid supporters say it works fine even with the variant. Critics say it doesn't, and you need methylfolate instead. Both sides are arguing about which form of folate to give to an enzyme that, in people with the TT genotype, is losing its grip on something else entirely.
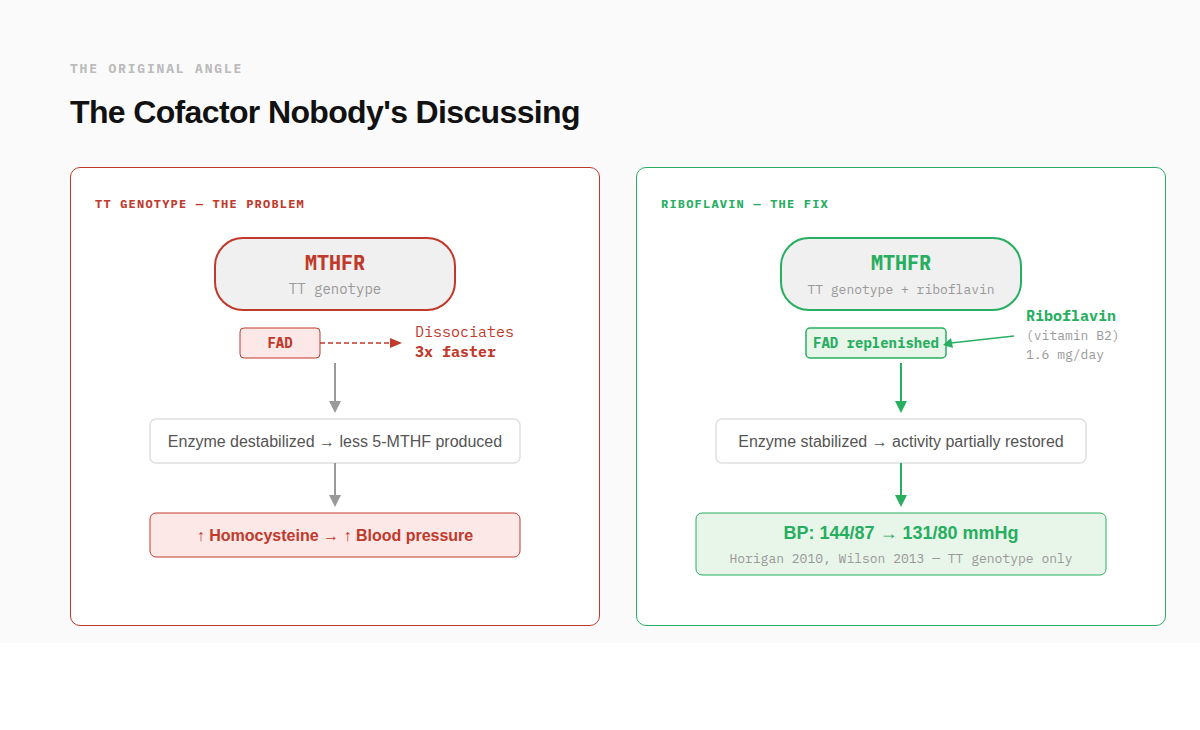
MTHFR is a flavoprotein. It requires a cofactor called FAD (flavin adenine dinucleotide) to function. The C677T variant doesn't just reduce enzyme activity in the abstract. It produces a "thermolabile" enzyme that loses its FAD cofactor three times faster than the “non-mutated” version of the enzyme.¹³ The enzyme isn't permanently broken. It's unstable. And the precursor to FAD is riboflavin: vitamin B2.
One of the best-documented consequences of reduced MTHFR activity is elevated homocysteine, which damages blood vessel lining and is consistently associated with hypertension. Multiple meta-analyses have confirmed that the TT genotype carries an independent increased risk of high blood pressure. So if riboflavin truly stabilizes the variant enzyme, blood pressure is exactly where you'd expect to see it show up first.
A research group at Ulster University has been testing this for over a decade. Horigan et al. (2010) ran a randomized, placebo-controlled trial in cardiovascular disease patients stratified by MTHFR genotype. Those with the TT genotype who received just 1.6 mg per day of riboflavin (that’s equivalent to the RDA lactating women) for 16 weeks saw blood pressure drop from 144/87 to 131/80 mmHg.¹⁴ The CT (remember, this is the single allele variant that drops enzyme activity by 30%) and CC (wild-type or “normal” function) groups showed no response. The effect was entirely genotype-specific.
Wilson et al. (2013) replicated it in hypertensive patients without overt cardiovascular disease: same dose, same genotype-specific result.¹⁵ A 4-year follow-up confirmed the effect persisted.¹⁶ And in 2020, Ward et al. analyzed over 6,000 adults and found that people with the TT genotype who also had low riboflavin status had a 3-fold increased risk of hypertension compared to those with the CC genotype and normal riboflavin.¹⁷
This is what personalized nutrition based on MTHFR variants actually looks like when it follows the evidence. Not a debate about which form of folate to take, but a different vitamin entirely, targeting the specific cofactor the variant enzyme can't hold onto, with replicated RCT data showing a clinically meaningful outcome in the genotype that's supposed to matter most.
And almost nobody in the MTHFR conversation mentions it.
What the Evidence Actually Supports
Folic acid works. Not theoretically, not in rodent models, but in the largest and most consequential supplement intervention in modern public health. Mandatory fortification reduced neural tube defects by 36% across all genotypes. Serum folate deficiency dropped from 30% to less than 1%. These results have held for over 25 years.
The MTHFR C677T variant is real, common, and measurable. Those with the TT genotype have roughly 30% of normal MTHFR enzyme activity, and their blood folate runs about 16% lower on the same intake. But "reduced" is not "broken." Folic acid raises blood folate in every genotype group, including TT. No randomized trial has demonstrated harm from folic acid at the recommended 400 mcg dose in any population, including those with the TT genotype.
The concerns about UMFA are biologically plausible but clinically unproven. The strongest trial in support of that argument (Cole 2007) used 2.5 times the recommended dose in a population already at elevated cancer risk, and wasn't replicated in combined analyses.⁹ The NIH's own review concluded that evidence of harm from UMFA remains inconclusive. Transport competition at the receptor level has been demonstrated in rare neurological conditions at therapeutic doses, not at fortification levels in the general population.
What is undersold: riboflavin. The one nutrient with replicated, genotype-specific, randomized trial evidence for a clinical outcome in people with the TT genotype. It targets the actual molecular defect, not the downstream product. And it barely registers in the conversation.
What's still missing is the study that would settle this for good: a large, pre-registered trial directly comparing folic acid, 5-MTHF, and riboflavin, stratified by MTHFR genotype, measuring both biochemical markers and clinical endpoints over several years. Until that trial exists, the strongest evidence-based position is also the simplest: you can take folic acid if you need it (that’s not the same thing as saying take over methylfolate or anything else. If you need folate, folic acid is better than no folate), make sure your B2 intake is adequate, and be skeptical of anyone who tells you a common gene variant makes a proven public health intervention dangerous.

References:
1. MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338(8760):131-137.
2. Williams J, Mai CT, Mulinare J, et al. Updated estimates of neural tube defects prevented by mandatory folic acid fortification — United States, 1995-2011. MMWR Morb Mortal Wkly Rep. 2015;64(1):1-5.
3. Pfeiffer CM, Hughes JP, Lacher DA, et al. Estimation of trends in serum and RBC folate in the U.S. population from pre- to postfortification using assay-adjusted data from the NHANES 1988-2010. J Nutr. 2012;142(5):886-893.
4. Bailey SW, Ayling JE. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc Natl Acad Sci U S A. 2009;106(36):15424-15429.
5. Pfeiffer CM, Sternberg MR, Fazili Z, et al. Unmetabolized folic acid is detected in nearly all serum samples from US children, adolescents, and adults. J Nutr. 2015;145(3):520-531.
6. Botto LD, Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151(9):862-877.
7. Akiyama T, Kuki I, Kim K, Yamamoto N, et al. Folic acid inhibits 5-methyltetrahydrofolate transport across the blood-cerebrospinal fluid barrier: clinical biochemical data from two cases. JIMD Rep. 2022;63(6):529-535.
8. Troen AM, Mitchell B, Sorensen B, et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J Nutr. 2006;136(1):189-194.
9. Cole BF, Baron JA, Sandler RS, et al. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA. 2007;297(21):2351-2359.
10. Figueiredo JC, Mott LA, Giovannucci E, et al. Folic acid and prevention of colorectal adenomas: a combined analysis of randomized clinical trials. Int J Cancer. 2011;129(1):192-203.
11. Maruvada P, Stover PJ, Mason JB, et al. Knowledge gaps in understanding the metabolic and clinical effects of excess folates/folic acid: a summary, and perspectives, from an NIH workshop. Am J Clin Nutr. 2020;112(5):1390-1403.
12. Crider KS, Zhu JH, Hao L, et al. MTHFR 677C→T genotype is associated with folate and homocysteine concentrations in a large, population-based, double-blind trial of folic acid supplementation. Am J Clin Nutr. 2011;93(6):1365-1372.
13. Yamada K, Chen Z, Rozen R, Matthews RG. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci U S A. 2001;98(26):14853-14858.
14. Horigan G, McNulty H, Ward M, Strain JJ, Purvis J, Scott JM. Riboflavin lowers blood pressure in cardiovascular disease patients homozygous for the 677C→T polymorphism in MTHFR. J Hypertens. 2010;28(3):478-486.
15. Wilson CP, McNulty H, Ward M, et al. Blood pressure in treated hypertensive individuals with the MTHFR 677TT genotype is responsive to intervention with riboflavin: findings of a targeted randomized trial. Hypertension. 2013;61(6):1302-1308.
16. Wilson CP, Ward M, McNulty H, et al. Riboflavin offers a targeted strategy for managing hypertension in patients with the MTHFR 677TT genotype: a 4-y follow-up. Am J Clin Nutr. 2012;95(3):766-772.
17.Ward M, Hughes CF, Strain JJ, et al. Impact of the common MTHFR 677C→T polymorphism on blood pressure in adulthood and role of riboflavin in modifying the genetic risk of hypertension: evidence from the JINGO project. BMC Med. 2020;18(1):318.